
Introduction
Oncocytomas are benign tumors composed of mitochondria-rich cells and can arise in organs such as the kidney, thyroid, salivary glands, and adrenal glands. While renal oncocytomas are relatively common, adrenal oncocytomas are rare, with less than 200 cases reported.1They typically affect women in their fifth decade and are usually located in the left adrenal gland. Although benign, their imaging often mimics adrenocortical carcinoma, making diagnosis challenging.2
We report the case of a 25-year-old man who presented a right hypochondrial pain, with examination revealing a right adrenal tumor.
Case report
A 25-year-old male with no significant medical history presented with a two-year history of right hypochondrial pain. Physical examination revealed stable vital signs and a non-tender tumor in the right upper quadrant.
Abdominopelvic computed tomography (CT) shows an inter-hepatorenal tumor measuring 18 cm in its longest axis. Further characterization with abdominal magnetic resonance imaging (MRI) revealed a well-defined, solid, oval adrenal mass measuring 17cm in height,13.5 cm transversely, and 13cm anteroposteriorly. The tumor appeared hypointense on T1-weighted images and iso to hyperintense on T2-weighted images, with intense progressive contrast enhancement without washout. Despite its large size, the mass remained well-demarcated from adjacent structures, though it was in broad contact with the posterolateral segment of the right liver and caused compression of the retrohepatic vena cava (Figure 1).
__coronal_(b_c)_t2-weighted_sequence__an.png)
Laboratory evaluation showed normal renal function (creatinine 7 mg/L), fasting glucose (0.9 g/L), and CEA (2.73 ng/mL). Hormonal tests confirmed a non-functioning tumor with normal levels of metanephrines (0.29 µmol/L), normetanephrines (0.8 µmol/L), cortisol (57.4 µg/24h), and catecholamines.
Considering the clinical, biochemical, and imaging findings, the differential diagnoses included adrenocortical carcinoma (ACC), pheochromocytoma, metastasis, and lipid-poor adenoma. Pheochromocytoma was excluded biochemically; the absence of a primary lesion argued against metastasis; and CT density/washout were indeterminate, leading to surgical resection for both diagnosis and treatment.
Given the tumor’s size and indeterminate imaging features, a right adrenalectomy was performed through a subcostal incision. Intraoperatively, the tumor was adherent to the upper pole of the right kidney and compressed the retrohepatic IVC. It was excised completely without rupture or complications. Oral intake resumed on day one; discharge followed on day four.
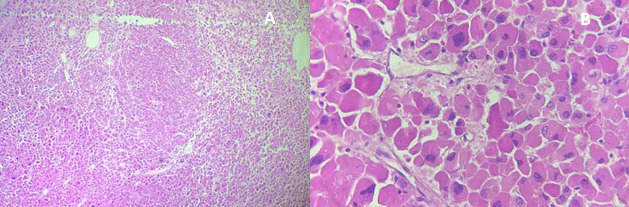
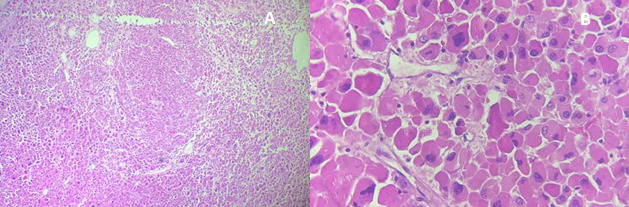
Histology revealed a well-circumscribed oncocytic adrenocortical neoplasm composed of large eosinophilic cells, without necrosis, mitoses, or venous, capsular, or sinusoidal invasion (Figure 2). The mitotic index was 0/50 HPF (≈0/10 mm²); Ki-67 labeling index was not assessed. Based on the Lin–Weiss–Bisceglia (LWB) system, the tumor met one minor criterion (size >10 cm) and no major criteria, classifying it as an adrenocortical oncocytic neoplasm of uncertain malignant potential (AONUMP), as recognized in the 2022 WHO classification .3,4 Immunohistochemistry was not performed, as morphology was unequivocally adrenocortical; in equivocal cases, a cortical panel (SF-1, inhibin-α, Melan-A, calretinin, often with synaptophysin co-expression) is recommended to confirm lineage and exclude mimics.5
The patient was followed at one, six, and twelve months, with no clinical or radiological evidence of recurrence.
DISCUSSION
Oncocytic tumors are composed of cells with abundant eosinophilic, granular cytoplasm due to mitochondrial accumulation.1The term “oncocytoma” refers to tumors made predominantly of these cells and commonly arises in the kidney, thyroid, and salivary glands. Adrenal oncocytomas are exceedingly rare, first described by Kakimoto et al. in 1986, with fewer than 200 cases reported.6 Adrenocortical oncocytic neoplasms (AONs) are rare and show female and left-sided predominance with diagnosis in mid-adulthood; therefore, a 25-year-old male with a right-sided mass is atypical.7 Most AONs are <10 cm; tumors ≥15 cm are uncommon and often mimic carcinoma radiologically. At 18 cm, this lesion qualifies as giant; it meets one LWB minor criterion (size >10 cm) with no major criteria and is therefore classified as AONUMP.3,4 WHO 2022 continues to endorse the LWB system: any major criterion denotes carcinoma; 1–4 minor criteria denote uncertain malignant potential; and absence of criteria denotes oncocytoma—our case had one minor criterion (size).4,5,8
Adrenal oncocytic tumors are usually benign and non-functioning, often discovered incidentally during imaging for unrelated issues.9However, hormonal activity is reported in up to 31.5% of cases, mimicking conditions like pheochromocytoma or Cushing’s syndrome.2,10,11Biochemical and clinical assessment is therefore essential, particularly in symptomatic or functional tumors. In our case, the diagnosis was made in the setting of abdominal pain, without evidence of hormonal excess.
CT and MRI lack specific features to reliably distinguish adrenal oncocytomas from malignancies. Typical findings include tumors >6 cm, lipid-poor content, and high attenuation (20–40 HU) on non-contrast CT.10 Unlike renal oncocytomas, they rarely exhibit a central scar.12 MRI often shows heterogeneous, iso- to hypointense T1 and hyperintense T2 signals, with variable diffusion restriction.12 Although features like fibrous encapsulation and heterogeneous enhancement may suggest malignancy, these are non-specific, complicating preoperative diagnosis.9,13,14
The diagnosis of adrenal oncocytoma is confirmed histologically using the Lin-Weiss-Bisceglia (LWB) system.15This system classifies tumors as malignant if any major criteria are present; high mitotic rate (>5/50 HPF), atypical mitoses, or venous invasion; and as borderline with one to four minor criteria, such as necrosis, large size, or capsular/sinusoidal invasion .15Tumors lacking all criteria are considered benign. In our case, histology revealed no mitotic activity, necrosis, or vascular or capsular invasion, consistent with AONUMP.4
Key pathological mimics include oncocytic pheochromocytoma/paraganglioma, characterized by a Zellballen architecture and immunoreactivity for chromogranin and synaptophysin with S100-positive sustentacular cells; these are typically SF-1 negative.11 Metastatic renal cell carcinoma can also mimic the morphology, usually expressing PAX8, CAIX, and CD10, but lacking SF-1. An oncocytic adrenocortical carcinoma (ACC) is distinguished by the presence of LWB major criteria and often a higher Ki-67 index. When required, SF-1 together with a cortical panel (inhibin-α, Melan-A, calretinin) reliably confirms adrenocortical origin.5 In contrast, renal oncocytoma often shows a central scar radiologically and expresses PAX8/CD117 (KIT), whereas adrenal oncocytoma lacks these renal lineage markers and is SF-1 positive.5,8 Oncocytomas of the thyroid and salivary glands instead express organ-specific markers, which helps exclude metastases to the adrenal.
Surgical excision is the treatment of choice for adrenal oncocytomas, with the approach-laparoscopic or open-based on tumor size, imaging features, and surgical expertise.6,16 Laparoscopy is preferred for small, well-encapsulated tumors due to lower morbidity and faster recovery13,14 and is feasible for lesions up to 12 cm. For tumors >8 cm, open adrenalectomy remains standard per ESMO guidelines.6,16 Outcomes are comparable between both techniques, allowing individualized selection based on anatomy and surgeon experience.6,12,16
Complete surgical resection offers excellent outcomes, with recurrence being exceptionally rare; only one case reported in the literature.10Although no standardized follow-up exists, most authors recommend annual imaging for 3–5 years, particularly for large or borderline tumors.12 In our case, CT scans at 1, 6, and 12 months showed no recurrence. Hormonal reassessment is unnecessary in non-functional, completely resected tumors unless symptoms appear.12Corticosteroid replacement is typically not required after unilateral adrenalectomy, as the contralateral gland maintains hormonal function.17
Conclusion
Adrenal oncocytomas are uncommon adrenal neoplasms that can mimic malignancy on imaging, especially when large. Histopathological examination remains essential for diagnosis and risk classification. This case underscores the importance of surgical excision for indeterminate adrenal masses and highlights the favorable outcome following complete resection of an adrenocortical oncocytic neoplasm of uncertain malignant potential.
Consent for publication
Written informed consent was obtained from the patient for publication and any accompanying images.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding
Not applicable
Acknowledgements
Not applicable