


Introduction
Acute thoracic aortic dissection (ATAD) is a catastrophic vascular condition associated with high morbidity and mortality, with estimates of 3–6 cases per 100,000 person-years and mortality rate approaching 50% within 48 hours without intervention.1,2 Heritable thoracic aortic diseases (HTAD), including Marfan syndrome, Loeys-Dietz syndrome, and vascular Ehlers-Danlos syndrome, predispose patients to aortic dissections at younger ages and often at smaller aortic diameters compared to sporadic cases.3 Loeys-Dietz syndrome, caused by pathogenic variants in TGFBR1 or TGFBR2, is characterized by aggressive arterial aneurysms and dissections, craniofacial anomalies, and systemic connective tissue involvement.4 Early recognition of these syndromes is essential, as prophylactic surgical intervention can prevent fatal complications.5
Imaging plays a pivotal role in both diagnosis and management of ATAD. CTA provides rapid, high-resolution evaluation of aortic morphology, extent of dissection, branch vessel involvement, and intramural hematoma.6 MRA offers excellent soft tissue contrast and is ideal for follow-up without radiation exposure.7 Despite advances in imaging, delayed recognition remains common, highlighting the need for high clinical suspicion, particularly in younger patients presenting with sudden chest or back pain and phenotypic signs suggestive of connective tissue disorders.8 In this case, we present a rare case of ATAD in a patient with previously undiagnosed Loeys-Dietz syndrome, emphasizing the integration of clinical, imaging, and genetic data to guide emergent surgical management and long-term follow-up.
Case Presentation
A 42-year-old male presented to the emergency department with sudden, severe tearing chest pain radiating to the back, accompanied by diaphoresis and nausea. He had no prior history of hypertension or cardiovascular disease. Upon examination, his blood pressure was elevated in both arms (180/95 mmHg in the right arm, 160/90 mmHg in the left arm), with a heart rate of 110 bpm. Physical features included mild pectus excavatum and disproportionately long extremities, suggestive of an underlying connective tissue disorder. Laboratory evaluation revealed a white blood cell count of 11,200/µL and a D-dimer of 2.3 µg/mL, while troponin I was within normal limits at 0.03 ng/mL (Table 1). Electrocardiography demonstrated sinus tachycardia without ischemic changes (Figure 1).
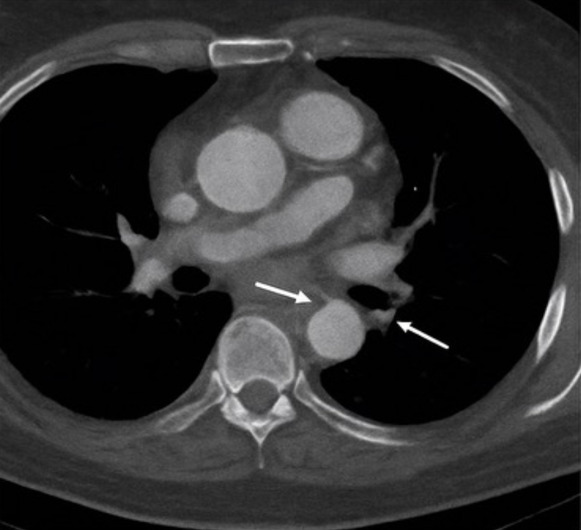
Emergent computed tomography angiography (CTA) revealed a Stanford type A aortic dissection originating in the ascending aorta and extending into the proximal descending thoracic aorta (Figure 2). The true lumen was patent, and no pericardial effusion was identified (Figure 3). The maximal diameter of the ascending aorta measured 6.2 cm. The dissection involved the aortic root and partially extended into the arch, while the innominate and left common carotid arteries remained patent. Transthoracic echocardiography revealed mild aortic regurgitation with preserved left ventricular ejection fraction (55%) and no pericardial effusion.

Based on imaging findings and clinical presentation, emergent surgical repair was indicated. The patient underwent ascending aortic replacement with a 26-mm Dacron graft and partial arch reconstruction under cardiopulmonary bypass with circulatory arrest at 25°C for 28 minutes. A median sternotomy was performed, and cardiopulmonary bypass was instituted via arterial cannulation of the proximal arch and bicaval venous cannulation. Total cardiopulmonary bypass time was 185 minutes, with an aortic cross-clamp time of 135 minutes.
Circulatory arrest was maintained for 28 minutes at 25°C with antegrade selective cerebral perfusion. Estimated blood loss was approximately 800 mL, requiring transfusion of 2 units of packed red blood cells and 1 unit of platelets intraoperatively. The ascending aorta and partial arch were replaced with a 26-mm Dacron graft, and the coronary buttons were reimplanted using the button technique. Intraoperative inspection revealed a thin, friable aortic wall, consistent with connective tissue disease. No intraoperative complications occurred.
Coronary reimplantation was performed using the button technique. Intraoperatively, the aortic wall was noted to be thin and friable, consistent with an underlying connective tissue disorder. No intraoperative complications occurred. Postoperatively, the patient remained hemodynamically stable, was extubated within 24 hours, and transferred to the intensive care unit for monitoring. The patient was transferred to the cardiovascular intensive care unit and remained hemodynamically stable. He was extubated within 24 hours and weaned off all vasoactive medications on postoperative day 1. He was mobilized on postoperative day 2 and transferred to the step-down unit on day 3. Postoperative transthoracic echocardiography demonstrated a well-seated graft, preserved left ventricular ejection fraction (55%), and only mild residual aortic regurgitation. No neurological, renal, or infectious complications occurred. The patient was discharged home on postoperative day 10 on a regimen of a beta-blocker and an angiotensin receptor blocker for blood-pressure and aortic protection.
Genetic testing revealed a heterozygous pathogenic TGFBR2 variant, confirming the diagnosis of Loeys-Dietz syndrome. Follow-up imaging with CTA and transthoracic echocardiography at 1, 6, and 12 months demonstrated stable grafts, no new aneurysms, and preserved ventricular function (Table 2). Mild residual aortic regurgitation was observed but was clinically insignificant (Table 3). The patient remained asymptomatic and was able to resume normal daily activities.
Discussion
Acute thoracic aortic dissection represents a high-stakes cardiovascular emergency, with rapid diagnosis and intervention critical for survival. HTADs, including Loeys-Dietz syndrome, confer increased susceptibility to dissection at younger ages and smaller aortic diameters than sporadic cases.9 Early recognition is therefore essential to guide both acute management and long-term monitoring.
Imaging remains central to diagnosis. CTA provides rapid and accurate assessment of aortic morphology, dissection extent, branch vessel involvement, and complications such as intramural hematoma or pericardial effusion.10 Echocardiography complements CTA by evaluating aortic valve function and left ventricular performance. MRA is especially useful for long-term follow-up, avoiding ionizing radiation.11 Prompt identification of the dissection type (Stanford type A vs. B) is essential, as type A dissections require emergent surgical intervention due to high mortality.12
Genetic evaluation plays an increasingly important role in patients with suspected HTAD. TGFBR1/TGFBR2 mutations in Loeys-Dietz syndrome are associated with aggressive aneurysm formation and early dissection, warranting vigilant surveillance and prophylactic repair.13 Multidisciplinary management, including cardiology, cardiothoracic surgery, radiology, and genetics, therefore essential to optimize outcomes.
Surgical repair remains the gold standard for type A dissection, with techniques including aortic root replacement, valve-sparing procedures, and arch reconstruction as indicated.14 Our patient underwent successful emergent repair, with stable postoperative outcomes confirmed on serial imaging. Long-term monitoring is essential, as patients with HTAD remain at risk for subsequent dissections and aneurysms. This case underscores the need for high clinical suspicion, rapid multimodal imaging, and integration of genetic data to guide management in rare HTAD-related aortic emergencies.
This case underscores the diagnostic and management challenges of acute type A dissection in heritable thoracic aortic disease (HTAD). Genetic confirmation of a pathogenic TGFBR2 variant established Loeys-Dietz syndrome (LDS) and explained the intraoperative arterial fragility, consistent with disrupted TGF-β signaling and medial degeneration.14 Genetic diagnosis also justifies lower thresholds for elective surgery, family screening, and lifelong surveillance.15
The patient underwent emergency ascending aortic replacement with partial arch reconstruction. Although valve-sparing techniques are reasonable in elective HTAD, the acute presentation and friable tissue favored a graft repair.16 Recovery was uncomplicated, with preserved ventricular function and only mild regurgitation at 12 months, consistent with registry data supporting improved survival following timely repair of type A dissections.17
While patients with Loeys–Dietz syndrome frequently dissect at smaller aortic diameters than sporadic cases (often prompting prophylactic repair when the aortic root reaches 4.0–4.5 cm depending on genotype, family history, and clinical features), our patient presented with an ascending aorta measuring 6.2 cm at the time of dissection, highlighting phenotypic heterogeneity within LDS and reinforcing the need for individualized surgical decision-making and vigilant lifelong surveillance.18 Literature describes both small-diameter dissections and rare large-root events, highlighting phenotype heterogeneity even within TGFBR2 cohorts.19,20 Mechanistically, abnormal TGF-β/SMAD signaling drives extracellular matrix remodeling, elastin fragmentation, and smooth muscle dysfunction, accounting for tissue fragility and explaining why dissections may occur at either small or large diameters.21 This biology underpins guideline recommendations for lower surgical thresholds and lifelong vascular imaging.22,23
Strengths of this report include multimodal diagnostic confirmation (clinical, imaging, genetic), genotype–phenotype correlation, and systematic follow-up confirming early surgical durability. Limitations include the single-patient nature and relatively short follow-up, as distal aortic events remain well-documented in LDS.24 Larger studies are needed to clarify whether large-diameter dissections represent exceptions or an underrecognized manifestation of LDS.25,26
Compared with sporadic ATAAD, which typically presents in the sixth decade, this case occurred at age 42, consistent with HTAD but older than many LDS cohorts, where dissections are often reported in the second to fourth decades.27,28 Overall, this case illustrates a rare large-diameter LDS dissection, reinforcing the importance of genotype-informed surveillance, tailored surgical strategies, and vigilant long-term follow-up to optimize survival.
Conclusion
Loeys–Dietz syndrome is a rare but highly lethal heritable thoracic aortic disease, characterized by early vascular vulnerability and a high risk of acute thoracic aortic dissection. Early detection by multimodal imaging is crucial to facilitate urgent surgical repair, particularly in Stanford type A dissections, where timely grafting and reconstruction can be lifesaving. Long-term management requires lifelong surveillance, genetic evaluation, and coordinated multidisciplinary care to improve survival and prevent recurrent vascular complications.